Streamlined Protein Structure Exploration: BioIn in PRinS3 Software Suite for Effortless Protein Refinement
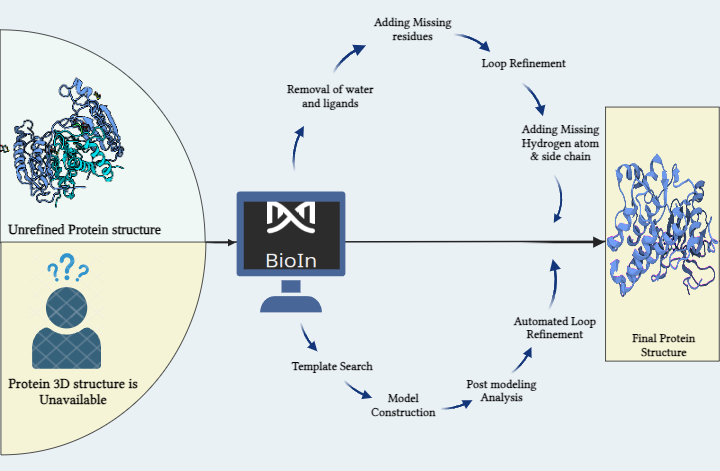
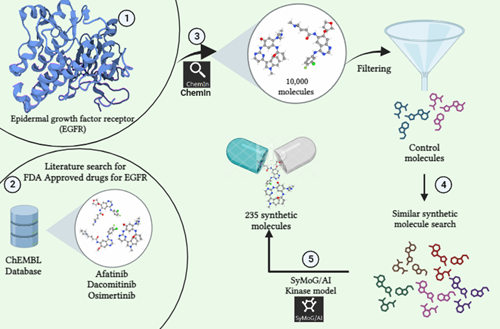
Commencing on the fascinating world of in silico studies, our journey begins with the exploration and identification of protein targets. Exploiting the power of databases like RCSB Protein Data Bank and Uniprot, we dive into the domain of protein structures. However, navigating the complexities faced in protein refinement and homology modeling is where the BioIn platform within PRinS3 software shines.
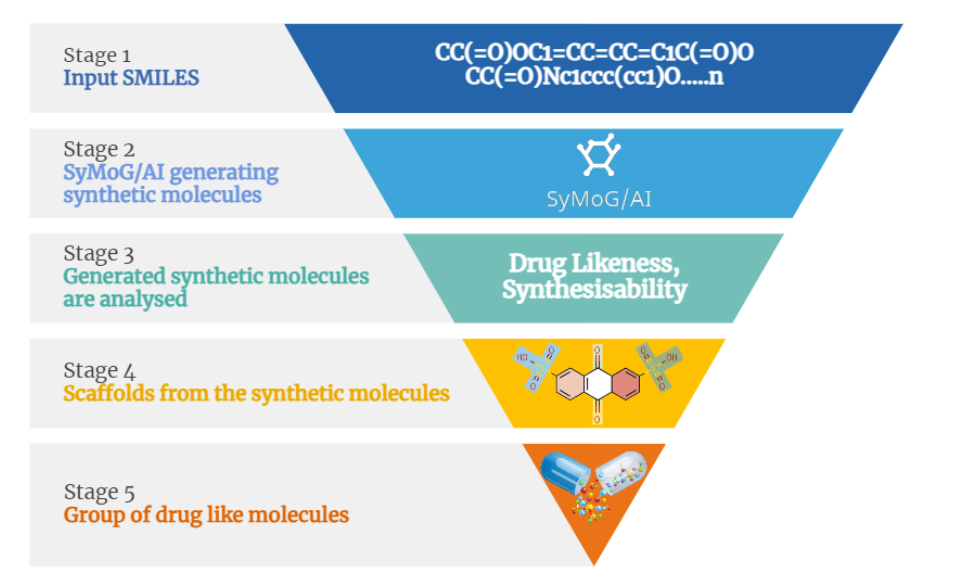
Within this innovative solution lies the key to overcoming the hustle in refining proteins with existing 3D structures and modeling those without. The process involves a sequence of steps: identifying the optimal structure, removing crystallization water and inhibitors, streamlining complex structures by eliminating unwanted chains, and refining through alignment, residue addition, loop refinement, and incorporation of missing atoms and side chains. This systematic refinement yields a fresh, analysis-ready protein structure, important for subsequent molecular docking and simulations. The significance of a 3D protein structure cannot be neglected in these calculations. Modeling stands tall as a crucial component in crafting this structure from an amino acid sequence. Here, we dive into the methods to prepare a protein for molecular docking, addressing both scenarios: when a protein structure is available and when it needs to be generated from scratch.
- Entering Scenario 1, we initiated on refining the protein using the BioIn application within PRinS3 software.Our focus was the target protein, Caspase 8, identified via the PDB ID (1QTN) fetched from RCSB PDB via BioIn. The analysis revealed missing residues, additional ligands, and inhibitors within the 3-chain protein structure, accompanied by water of crystallization. Notably, 17 residues were absent from the structure, prompting our first step: removing the ligand and crystallization water. Subsequently, the protein underwent enhancement within BioIn's structure editor. Missing residues were strategically added by aligning the structure with a template via NCBI BLASTp search, eliminating any gaps in the modeled structure. Further refinement ensued through loop refinement, generating 5 models, among which the one with the lowest dope score (-546.57928) was selected. This model was then fortified with hydrogen atoms and the missing side chain. This systematic series of carefully executed steps resulted in a refined, immaculate protein structure, poised for the next stage: molecular docking.
- Scenario 2: Imagine exploring into a project where the protein's 3D structure was an mystery, and the solution lay in homology modeling using BioIn within the PRinS3 software suite.To unveil the three-dimensional structure of the Alpha-1B adrenergic receptor (PDB ID: 7B6W) through the art of homology modeling. The journey began with the FASTA sequence of the protein, our gateway to unraveling its structure.
The main step is the template selection. We scoured the Protein Data Bank (PDB) for proteins, deploying tools like BLAST and FASTA to find the ideal template. It was crucial to find a template that resembles our protein's structure with a significant 95% similarity. Visualizing this journey was a dendrogram on the BioIn window, a map displaying identity and R-Factor values, guiding us to the best template.Once we finalized our template, aligning the sequences was paramount. Refined alignment algorithms were utilized by the platform, generating an optimal alignment, yielding specific alignment scores a critical phase before initiating the model construction. Post-modeling, the window revealed statistical scores DOPE and GA341 keys to gauging energy levels. Models with lower DOPE or higher GA341 scores signified more stable energy. Among the array of models, PI12.B99990002.pdb stood out, our take away for further analysis.
Evaluating the model was next an extensive scan for missing residues, side chains, and steric clashes. BioIn presented a graph plotting DOPE Per residue scores, spotlighting the conserved regions, bridging the model and template.
In the realm of sequence alignment, gaps emerged insertions and deletions calling for loop modeling. The automated loop refinement, a seamless process within the application, yields a set of refined models, each carrying its mol pdf values energy in numerical form.
The search for the optimal model continued, seeking the lesser positive values, signifying a model's goodness. We have reached the end point with our final structure PI12.BL00030001.pdb the end result of a well-structured model for our protein.
The journey reached a conclusion in a fully structured 3D protein, carefully crafted within BioIn, a testament to the power of a single application.